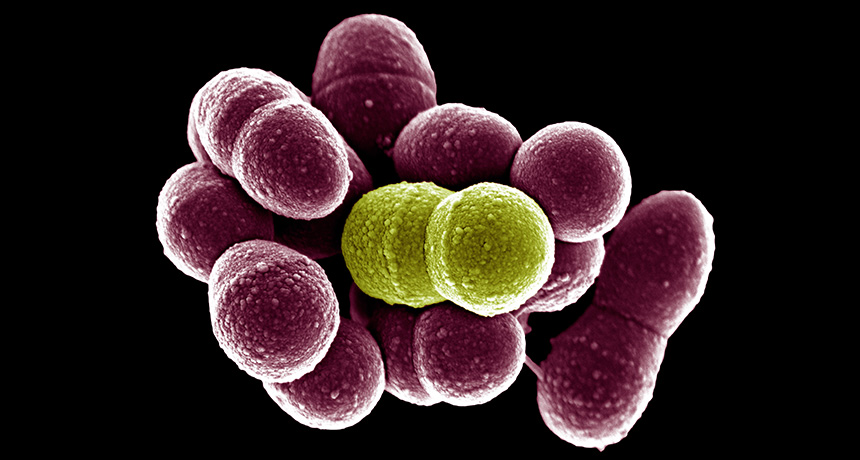
Normally harmless mouth bacteria can be a bad influence. When they pal around with tooth- and gum-attacking microbes, they can help those pathogens kick into high gear. This teamwork lets infections spread more easily — but also could offer a target for new treatments, scientists report online June 28 in mBio.
The way that bacteria interact with each other to cause disease is still poorly understood, says study coauthor Apollo Stacy, of the University of Texas at Austin. Lab work often focuses on individual bacteria species, but managing the communities of microbes found in living organisms is a more complex task. The new finding suggests that bacteria can change their metabolism in response to the presence or absence of other bacteria. A benign species of bacteria excretes oxygen, allowing the second species to switch to a more efficient aerobic means of energy production and helping it become a more robust pathogen. This is the first time a normally harmless mouth bacterium has been shown to change a pathogen’s metabolism to make the microbe more dangerous, says Vanessa Sperandio, a microbiologist at University of Texas Southwestern Medical Center in Dallas who wasn’t part of the study. Similar interactions have been shown between gut bacteria, she says, but “it’s a very new field of research and there are very few examples.”
Stacy and his collaborators examined the relationship between two species of bacteria that tend to grow in the same place in the mouth. One, Streptococcus gordonii, is found in healthy mouths and only occasionally causes disease. The other, Aggregatibacter actinomycetemcomitans, frequently causes aggressive tooth and gum infections.
The researchers knew from previous work that the pathogenic bacteria grew better with a coconspirator. To figure out why, Stacy says, “we asked, ‘What genes do they need to live when they’re by themselves?’” The team compared the solo-living genes to those active in the pathogen while growing alongside S. gordonii. The analysis revealed that the pathogenic bacteria switched their metabolism when S. gordonii was around.
When A. actinomycetemcomitans grew alone, they produced energy without using oxygen — a slow way to grow. But with S. gordonii nearby, the pathogenic bacteria took advantage of the oxygen released by their neighbors and increased their energy production. When tested in mice, that increased energy let the pathogen grow faster and survive better in a wound.
The findings are part of a growing body of research showing that bacteria can sense the presence of other bacteria and adjust their behavior accordingly. “This system has allowed us to begin to understand that microbes are really astute at evaluating this biochemistry, and in response, they have very specific behaviors,” says Marvin Whiteley, a microbiologist at the University of Texas at Austin who worked with Stacy.
Stacy plans to test the phenomenon in other bacteria pairs to see whether it holds up beyond these two species. Understanding the way bacteria interact with each other could let doctors target infections more efficiently, he says. For instance, if a bacterial infection isn’t responding to antibiotic treatment, targeting the sidekick bacteria might help take the primary pathogen down.
It’s time for a final farewell to the comet lander Philae.
The European Space Agency announced that on July 27 it would shut off the equipment that the Rosetta spacecraft uses to listen in on communications from Philae. The lander, which touched down on comet 67P/Churyumov-Gerasimenko in November 2014, briefly transmitted data before entering a deep slumber.
Except for a brief awakening in June and July 2015, Philae has been silent ever since. Now, as the solar-powered Rosetta gets farther from the sun, scientists need to conserve power by shutting off nonessential equipment. So Rosetta will listen no more.
Rosetta will continue scientific operations around comet 67P for another two months before completing its mission, when it will join Philae, descending down onto the comet.
Activity of a gene that lowers levels of caffeine-degrading enzymes in the liver is associated with how much coffee people drink, researchers say August 25 in Scientific Reports. The more active the gene, called PDSS2, the less coffee people drank.
Researchers tracked the coffee-drinking habits of 1,207 people in remote Italian villages and 1,731 people from the Netherlands. The researchers looked for an association between sipping java and people’s genetic makeup. The Dutch quaffed, on average, more than five cups of filtered coffee per day; the Italians sipped about two cups of espresso. In the Italians, 21 genetic variants in DNA surrounding the PDSS2 gene were linked to coffee consumption, Nicola Pirastu, of the University of Edinburgh, and colleagues found. The strongest-acting variant changed espresso consumption by 1.09 cups per day. Only five of the variants found in the Italians seemed to alter coffee-drinking in Dutch people, and did so to a lesser extent.
Given the larger size of the cups, Dutch people consume about three times as much caffeine per cup as the Italians do. Other caffeine-processing genes, such as CYP1A2 (SN Online: 4/8/11), may control coffee consumption habits at higher caffeine doses, while PDSS2 limits low-level caffeine intake, the researchers speculate.
A new map of the galaxy, the most precise to date, reveals positions on the sky for over 1 billion stars both within and beyond the Milky Way.
This new galactic atlas, courtesy of the European Space Agency’s Gaia spacecraft, also provides distances to roughly 2 million of those stars, laying the groundwork for astronomers who want to piece together the formation, evolution and structure of the Milky Way.
“This is a major advance in mapping the heavens,” Anthony Brown, an astrophysicist at Leiden University in the Netherlands, said September 14 at a news briefing. “Out of the 1 billion stars, we estimate that over 400 million are new discoveries.” There are no major cosmic revelations yet; those will develop in the months and years to come as astronomers pore over the data. This catalog of stars is just a first peek at what’s to come from Gaia, which is spending five years gathering intel on a wide variety of celestial objects.
The final survey will eventually provide a 3-D map of over 1 billion stars. It will also chart positions of roughly 250,000 asteroids and comets within the solar system, 1 million galaxies, and 500,000 quasars — the blazing cores of galaxies lit up by gas swirling around supermassive black holes. Mission scientists also expect they will turn up over 10,000 undiscovered planets orbiting other stars.
“It’s a very democratic mission,” said project scientist Timo Prusti. “Anything that looks like a [point of light] gets caught up and observed.”
Gaia launched on December 19, 2013, and eventually settled into its home about 1.5 million kilometers from Earth on an orbit that follows our planet around the sun (SN Online: 12/19/13). Regular science observations started in July 2014. This first data release, described in a series of papers being published online starting September 14 in Astronomy & Astrophysics, contains data obtained through September 2015.
The spacecraft repeatedly scans the sky with two telescopes pointed in different directions. To make the 3-D map, Gaia measures each star’s parallax, a subtle apparent shift in the position of the star caused by the changing viewing angle as the spacecraft loops around the sun. By measuring the amount of parallax, and knowing the size of Gaia’s orbit, astronomers can triangulate precise distances to those stars. With distances in hand, astronomers can figure out how intrinsically bright those stars are, which in turn will help researchers understand how stars evolve. A detailed stellar map could also help chart the Milky Way’s distribution of dark matter, the elusive substance that is thought to make up the bulk of the mass in all galaxies and reveals itself only through gravitational interactions with stars and gas.
One controversy that astronomers are eager to resolve with Gaia is the distance to the Pleiades star cluster, one of the closest repositories of youthful stars. A previous Gaia-like mission, the Hipparcos satellite, came up with a distance of about 392 light-years. Estimates based on simulations of how stars evolve as well as observations from the Hubble Space Telescope and ground-based radio observatories pin the Pleiades at about 443 light-years away (SN Online: 4/28/14).
“Clusters give you a sense of the evolution of stars at different ages,” says Jo Bovy, an astrophysicist at the University of Toronto who is not involved with the Gaia mission. “The Pleiades is a nearby cluster that we can study well — it’s one of the cornerstones.” All of the stars in the Pleiades are roughly 100 million years old, and so provide a snapshot of a relatively young stellar age. Nailing down the distance reveals the intrinsic brightness of the stars, which is crucial to understanding how stars develop in their early years.
Gaia appears to be leaning toward the larger distance, Brown said, but there’s still too much uncertainty in the data to say anything definitive. “It’s too early to say how the controversy will be resolved. But Gaia will pin it down.”
The Pleiades distance debate, as well as a clearer picture of how the Milky Way is put together, will have to wait for future data releases from Gaia. The next release is planned for late 2017; the final catalog won’t be available until 2022.
“Those will be much more interesting,” Bovy says. “Then we can actually start using our modeling machinery and see how stars are distributed throughout the galaxy. We can test our understanding of dark matter and our understanding of how the Milky Way formed.”
Michael Snyder’s genes were telling him that he might be at increased risk for type 2 diabetes. The Stanford University geneticist wasn’t worried: He felt healthy and didn’t have a family history of the disease. But as he monitored other aspects of his own biological data over months and years, he saw that diabetes was indeed emerging, even though he showed no symptoms.
Snyder’s story illustrates the power of looking beyond the genome, the complete catalog of an organism’s genetic information. His tale turns the genome’s one-dimensional view into a multidimensional one. In many ways, a genome is like a paper map of the world. That map shows where the cities are. But it doesn’t say anything about which nations trade with each other, which towns have fierce football rivalries or which states will swing for a particular political candidate. Open one of today’s digital maps, though, and numerous superimposed data sources give a whole lot of detailed, real-time information. With a few taps, Google Maps can show how to get across Boston at rush hour, offer alternate routes around traffic snarls and tell you where to pick up a pizza on the way.
Now, scientists like Snyder are developing these same sorts of tools for biology, with far-reaching consequences. To figure out what’s really happening within an organism — or within a particular organ or cell — researchers are linking the genome with large-scale data about the output of those genes at specific times, in specific places, in response to specific environmental pressures. While the genome remains mostly stable over time, other “omes” change based on what genes are turned on and off at particular moments in particular places in the body. The proteome (all an organism’s proteins) and the metabolome (all the metabolites, or small molecules that are the outputs of biological processes) are two of several powerful datasets that become more informative when used together in a multi-omic approach. They show how that genomic instruction manual is actually being applied. “The genome tells you what can happen,” says Oliver Fiehn, a biochemist at the University of California, Davis. The proteome and the metabolome can show what’s actually going on.
And just as city planners use data about traffic patterns to figure out where to widen roads and how to time stoplights, biologists can use those entwined networks to predict at a molecular level how individual organisms will respond under specific conditions.
By linking these layers and others to expand from genomics to multi-omics, scientists might be able to meet the goals of personalized medicine: to figure out, for example, what treatment a particular cancer patient will best respond to, based on the network dynamics responsible for a tumor. Or predict whether an experimental vaccine will work before moving into expensive clinical tests. Or help crops grow better during a drought.
And while many of those applications are still in the future, researchers are laying the groundwork right now.
“Biology is being done in a way that’s never been done before,” says Nitin Baliga, director of the Institute for Systems Biology in Seattle.
Data dump Scientists have long studied how genes influence traits. Researchers have figured out important connections between genes and the proteins they encode and have scoured the genome for associations between particular genetic mutations and diseases. But a gene-by-gene view of the body is like trying to diagnose a citywide traffic problem by looking at just one backed-up intersection.
“There are so many places that a system can go awry,” Baliga says. When dozens of genes are working together, it’s tricky to tease out which one is misfiring in a particular instance.
Baliga is among a growing group of scientists who want to study life through a systems lens, because sometimes that traffic jam at one intersection is being caused by an out-of-sight accident three blocks away.
Such an approach is particularly useful for unraveling the complexities of diseases like cancer and diabetes. These conditions involve a tangled web of genes, paired with lifestyle factors and environmental conditions — Is she a smoker? How much does she exercise? — that influence when those various genes are turned on and off. Reconstructing the tangled routes by which genes interact to influence the body is a slightly more complicated feat than mapping the best path from Tulsa to Tuscaloosa. For one thing, it requires serious computer power to gather, store and analyze all that data. The 3 billion chemical coding units that string together to form a person’s inventory of DNA, if entered into an Excel spreadsheet line-by-line, would stretch about 7,900 miles. The human proteome contains more than 30,000 distinct proteins that have been identified so far. And researchers have cataloged more than 40,000 different human metabolites, such lactic acid, ethanol and glucose.
Working with such big datasets can be expensive, too. Compiling the first human genome took 10 years and cost almost $3 billion. Now, the costs of collecting and analyzing all these datasets have come down, so it’s finally feasible to use them in tandem to answer big biological questions.
The important players Scientists would like to understand the interplay between the genome and the proteome. Add in the metabolome. To make things more complex, there’s the epigenome — the chemical modifications to DNA that help control which genes are turned on and off — and the transcriptome, the full range of RNAs that translate DNA’s blueprints so they can be used to make proteins. It’s no surprise that mapping such a comprehensive network for any organism is still a distant goal.
For now, scientists tend to focus their multi-omic studies on a particular disease or question. Baliga wants to learn how tuberculosis — which sickens nearly 10 million people per year and kills 1.5 million — evades drugs within the body. Many strains of the TB bacterium are resistant to existing treatments or can tolerate them long enough to establish a strong foothold.
To learn how Mycobacterium tuberculosis mounts a defense against a drug, Baliga is first looking within the bacterium, identifying the genes, proteins and other molecules that interact as the microbe infects a host.
He collects different types of omic data from M. tuberculosis alone and when it’s in the presence of an antibiotic. His team recently focused on the microbe’s response to bedaquiline, a drug used to treat multidrug-resistant TB. Baliga measured the microbe’s transcriptome in the presence of different doses of bedaquiline and at different times after introducing the drug.
From this giant data dump, computer models helped narrow the focus to a smaller collection of genes, proteins and other molecules that changed under certain conditions. Visualization programs turned these mathematical outputs into maps that scientists could analyze.
About 1,100 genes behaved differently in the presence of bedaquiline, Baliga’s team reported in August in Nature Microbiology. Measurements of the RNA indicated that most of those genes became less active, but a few shifted into overdrive. The researchers suspected those hyperactive genes might be behind the resistance — playing off each other to create a smaller network within the larger tuberculosis response network.
But statistical analysis alone wasn’t enough to confirm the hunch. Correlation isn’t cause, Fiehn points out. Scientists need to figure out which of those changes actually matter. That is, if you’re scanning millions of data points looking for variation, you’re going to find certain abnormalities that are due to chance and are unrelated to the disease or question at hand. But starting from that smaller dataset of outputs that change, scientists can then test which players are actually important in the network and which belong on the sidelines. In animal models or petri dishes, scientists disable one gene at a time to see how it affects the proposed network.
“Systems biology has been able to generate these amazing hypotheses about how genes interact,” Baliga says. Testing them has historically been more challenging. But now, gene-editing technologies such as CRISPR/Cas9 (SN: 9/3/16, p. 22) allow scientists to more easily test these networks in living systems.
Baliga and his team edited the genome of M. tuberculosis, disabling the regulatory machinery responsible for some of the overactive genes. Sure enough, the drug worked better on the modified bacteria, the researchers reported.
Networking solutions Once a network has been mapped, scientists can use it to predict (and maybe even prevent) illness.
Baliga’s team identified a second drug that works with bedaquiline. The drug turns off some of the regulators for the overactive tuberculosis gene network that was fighting off the bedaquiline. Using the second drug with bedaquiline made tuberculosis bacteria more vulnerable, pointing to a potential strategy for dealing with persistent infections.
Baliga’s group is also mapping networks in patients with glioblastoma, a particularly deadly type of brain tumor. In the August Cell Systems, the scientists described work designed to figure out why some patients respond to certain drugs while others don’t. The aim is to personalize treatments, to choose a drug that targets the particular network glitch that gives rise to that patient’s tumor, Baliga says. The drug might ramp up production of a protein that’s currently in short supply, or turn off a gene that’s mistakenly on. That same drug might be completely useless for another patient whose tumor developed through a different network error.
“Being able to do that systematically across all cancers, using networks — that has not happened yet,” Baliga says. But scientists have devised drug treatments to address individual mutations. And expanding that to a greater range of cancers in the future is not farfetched, he says.
Other scientists are using multi-omic approaches for preventive medicine, for example, to be more effective and efficient in vaccine development. Despite years of trying, scientists still haven’t created an HIV vaccine that can protect people against the virus, says Alan Aderem, a biologist at the Center for Infectious Disease Research in Seattle. Bringing a vaccine from test tube to market is costly and time-consuming. With a better understanding of how the networks of the body’s immune system respond to the disease, researchers could be more selective in the vaccine candidates that they invest time and money in developing.
“The real value of systems biology is that it’s predictive,” Aderem says. “If you could predict upfront if a vaccine would work, you’d not only save a huge amount of energy, you’d also save a huge amount of lives.”
Plant power Multi-omics has perhaps received the most attention in the context of human health — but that’s also the realm where it’s hardest to piece together the omic layers. Because simpler organisms can be manipulated genetically, it’s easier to move from networks on a computer screen to real solutions. For instance, some scientists are using multi-omic analysis to engineer networks that let crop plants thrive with less water. To turn carbon dioxide into sugar via photosynthesis, plants need a lot of water. But some desert plants make do with less. Most plants take in carbon dioxide through small pores in their leaves. Opening these pores can let water evaporate out. In the desert, where water is in short supply, some plants use a different network of chemical reactions to make energy: crassulacean acid metabolism, or CAM.
Plants that use CAM open pores in their leaves only during the cooler nighttime, when water is less likely to evaporate out. They store the CO2 they take in until daytime, when they close the pores and convert the CO2 into food.
“Our goal is to move this metabolic trick into crop plants,” says John Cushman, a biochemist at the University of Nevada, Reno. “In order to do that, you have to understand the complexity of all the enzymes and regulatory components associated with the circadian clock of the plant.”
Cushman’s team has collected vast omic datasets from CAM plants. He estimates that several hundred genes coordinate the process, turning each other on and off and producing proteins that interact with each other and influence other genes.
His team is trying to engineer the plant Arabidopsis (a weed often used for genetic experiments) to use CAM by inserting the relevant genes into the plant. If they can get it to work in this small lab plant, the researchers want to do the same in poplar trees to help grow biofuel sources in harsh environments not usually suitable for agriculture. Someday, Cushman hopes, the technology will help food crops grow better in arid climates.
Better than a hunch So far, collecting and integrating omic data is largely restricted to the lab — the work is still expensive and time-consuming. But scientists like Snyder, who diagnosed himself with type 2 diabetes, think that someday people could regularly collect these sorts of data to track changes in their health over time.
The Stanford geneticist began by analyzing his own genome. Then he started measuring his proteome, metabolome and other omic data at regular intervals to develop what he termed his “personal omics profile.”
Certain mutations in his genome suggested that he was at risk for developing diabetes from the get-go. He was surprised, but didn’t take action. Nearly a year into his experiment, though, changes in his other omic data suggested that his body was no longer using glucose properly. His blood sugar was elevated and so was a type of hemoglobin that increases with uncontrolled diabetes, according to a report in Cell by Snyder and three dozen colleagues in 2012. Those aren’t new measures — they’re exactly what a doctor would test in someone with diabetes symptoms. But Snyder had no symptoms. He had just had a nasty cold, which he now thinks may have triggered the onset of diabetes because he was already genetically at risk.
Because he spotted the changes early and his doctor confirmed he had diabetes, Snyder was able to fend off symptoms by changing his diet and exercise habits — which was reflected in the follow-up data he collected.
He monitored how other biological measurements (molecules that standard medical diagnostic tests might not even look at) changed with the onset of his diabetes and the lifestyle changes he made in response. That information might be valuable for doctors hoping to detect the onset of diabetes in patients before symptoms appear. So now his lab is tracking omic data from 100 people, most of whom have elevated blood sugar levels, but have not yet been diagnosed with diabetes. Snyder wants to see whether viral infections trigger diabetes in other people, or whether his case is an isolated incident.
Snyder is still tracking fluctuations in his own data over time, too. He thinks it’s a powerful tool for personalized medicine because it shows in real time how an individual responds to disease and other stressors.
“You’ll be able to start linking disease to treatment to outcomes,” Snyder says. “It becomes what I call data-driven medicine, instead of hunch-driven medicine.” Making sense of this sort of data isn’t easy. “No story is the same,” Snyder says. “We see a lot of strange stuff that we don’t know how to interpret yet.”
But collecting detailed omic-level data about individuals over time could still be useful, he says. By collecting more data from healthy people, doctors and scientists could get a better sense of what’s normal. And tracking fluctuations in individual patients’ data over time can show what’s typical for that particular person. Snyder thinks that might be more valuable than knowing what’s normal for people in general. Someone monitoring these biological signs might notice molecular changes — as he did — before they cause troublesome physical symptoms.
For the multi-omic future that Snyder and others envision, scientists will need a greater ability to store and wrangle massive amounts of data than they possess today. And they’ll need ways to test the networks they uncover. But the potential payoff is enormous: A response map that can display the intertwining routes between genes and outcomes, as well as how distant red lights, speed bumps and construction zones play off each other to shift those paths.
Electron microscopy is finally getting its Kodachrome moment.
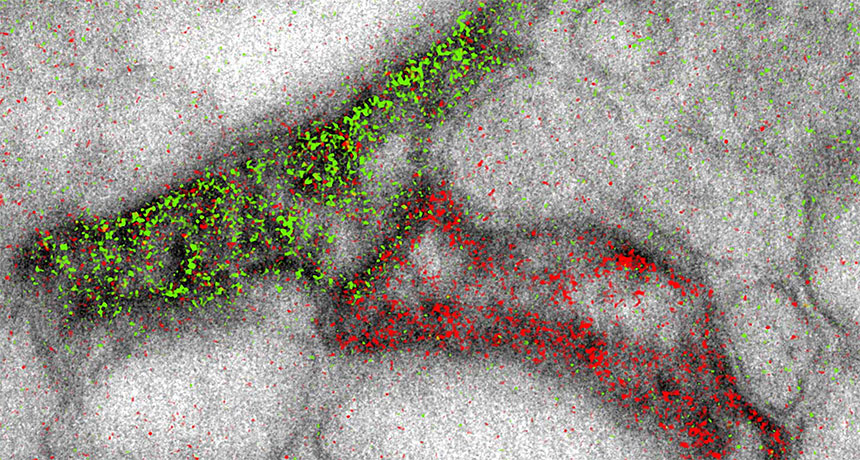
The high-powered scopes can now produce images that simultaneously highlight different molecules in different colors, scientists report online November 3 in Cell Chemical Biology. That’s helpful for researchers hoping to visualize the complex structures of cells or tissues — such as connections between brain cells, shown here.
Electron microscopes build black-and-white images by shooting beams of electrons at samples. Previously, scientists could add color by overlaying lower-resolution images from light microscopes. The new technique adds pizzazz without sacrificing image quality. It involves sequentially layering different metal ions on top of the sample. Each ion selectively latches onto a different target molecule. The electron beam interacts differently with each ion, yielding signature wave shapes that can be converted into colors. The researchers used the coloring technique to show that two brain cells called astrocytes (the edge of one shown in green, the other in red) could link up to the same message-sending junction between nerve cells.
A person growing up in Peru in the 1970s or 1980s probably didn’t eat anchoveta, the local species of anchovies. The stinky, oily fish was a food fit only for animals or the very poor. The anchoveta fishery may have been (and still is, in many years) the world’s largest, but it wasn’t one that put food on the table.
For thousands of years, though, anchoveta fed the people of Peru. It was only when the industrial fishing fleet got started in the 1950s — one that converted most of its catch into fishmeal for feeding other animals — that people lost interest in the fish.
In fact, of the some 20 million metric tons of fish caught annually around the world for uses other than eating, 90 percent are fish that are perfectly good to eat, such as sardines, anchovies and herring, a new study finds.
“Historically, these fish were eaten for human consumption,” but at some point, like the anchoveta, they gained a reputation as being “trash,” notes Tim Cashion, the study’s lead author.
Cashion is a researcher for the Sea Around Us project at the University of British Columbia’s Institute for the Ocean and Fisheries, and he and his colleagues have been collecting data to illuminate the impact of fisheries on marine ecosystems. Cashion spent a year tallying up fishery catches around the world from 1950 to 2010, figuring out who caught which species and where the fish went after it was taken out of the sea.
About 27 percent of ocean fish caught became fishmeal or fish oil. Those products were used to feed farmed fish or used in agriculture, for purposes such as feeding pigs and chickens. And most of the fish species were either food-grade (used as food somewhere in the world) or prime-food-grade (widely accepted as food everywhere), Cashion and his colleagues report February 13 in Fish and Fisheries.
“There could be a better use of these fish,” says Cashion. Instead of feeding fish to fish, the fish could feed people, especially those who lack access to high-quality sources of protein. But the reason why that doesn’t happen is a combination of economics and regulations. In many places, and for many fish, a fisherman can get more money for his catch if he sells it for fishmeal than if he sells it to the locals for their meals. And in Peru, those economic incentives have combined with a 1992 law and more-recent regulations to keep anchoveta off locals’ plates — despite increased demand brought on by efforts to change the fish’s poor reputation. (These included segments on a popular TV food show.)
By law, the industrial fishing fleet in Peru can only sell anchoveta for fishmeal. Only smaller, artisanal fleets can catch anchoveta for people to eat and can’t legally sell their catch for fishmeal — but the higher price offered by the fishmeal plants means that many do. As a result, the amount of anchoveta that people in Peru consumed dropped by more than half from 2011 to 2014, despite more people actually wanting to eat the fish.
But a change in how the anchoveta are handled could satisfy both the need to feed the Peruvian people and supply the fishmeal industry, Santiago de la Puente of the Institute for the Oceans and Fisheries and colleagues note February 15 in Fish and Fisheries. A ton of anchoveta caught for canning yields 150 kilograms of fish in the can and 600 kilograms of heads, guts and other bits that are perfect for fishmeal. If all the anchoveta were diverted through the canning industry, the researchers calculate a net yearly benefit of 40,000 jobs, $351 million in additional revenue and 40 kilograms of nutrient-rich, tasty fish for every person in Peru.
The 180-kilometer-long crack threatening one of Antarctica’s largest ice shelves has branched out, new satellite observations reveal. The main rift in the Larsen C ice shelf hasn’t grown longer since February. But radar mapping shows that a crack has split off from the main rupture like a snake’s forked tongue, members of Project MIDAS, an Antarctic research group, report May 1. That new branch, about 15 kilometers long, wasn’t on radar maps taken six days earlier, the team says.
If either branch makes it to the edge of Larsen C, the shelf could calve off a 5,000-square-kilometer hunk of ice (SN: 7/25/15, p. 8), creating one of the largest icebergs ever recorded, says glaciologist Adrian Luckman of Swansea University in Wales. “The new branch is heading off more toward the ice front, so it’s more dangerous and more likely to cause this calving event to occur” than the main branch, he says. Snapping off such a large ice chunk could destabilize the entire ice shelf, Luckman warns. A similar event led to the collapse of Larsen B in 2002. Because Larsen C’s ice floats on the ocean, the loss wouldn’t directly raise sea levels. But its demise could serve as a case study of how other shelves may break apart as rising temperatures melt and weaken Antarctic ice, Luckman says.
You’ve probably encountered at least one machine-learning algorithm today. These clever computer codes sort search engine results, weed spam e-mails from inboxes and optimize navigation routes in real time. People entrust these programs with increasingly complex — and sometimes life-changing — decisions, such as diagnosing diseases and predicting criminal activity.
Machine-learning algorithms can make these sophisticated calls because they don’t simply follow a series of programmed instructions the way traditional algorithms do. Instead, these souped-up programs study past examples of how to complete a task, discern patterns from the examples and use that information to make decisions on a case-by-case basis. Unfortunately, letting machines with this artificial intelligence, or AI, figure things out for themselves doesn’t just make them good critical “thinkers,” it also gives them a chance to pick up biases.
Investigations in recent years have uncovered several ways algorithms exhibit discrimination. In 2015, researchers reported that Google’s ad service preferentially displayed postings related to high-paying jobs to men. A 2016 ProPublica investigation found that COMPAS, a tool used by many courtrooms to predict whether a criminal will break the law again, wrongly predicted that black defendants would reoffend nearly twice as often as it made that wrong prediction for whites. The Human Rights Data Analysis Group also showed that the crime prediction tool PredPol could lead police to unfairly target low-income, minority neighborhoods (SN Online: 3/8/17). Clearly, algorithms’ seemingly humanlike intelligence can come with humanlike prejudices.
“This is a very common issue with machine learning,” says computer scientist Moritz Hardt of the University of California, Berkeley. Even if a programmer designs an algorithm without prejudicial intent, “you’re very likely to end up in a situation that will have fairness issues,” Hardt says. “This is more the default than the exception.” Developers may not even realize a program has taught itself certain prejudices. This problem gets down to what is known as a black box issue: How exactly is an algorithm reaching its conclusions? Since no one tells a machine-learning algorithm exactly how to do its job, it’s often unclear — even to the algorithm’s creator — how or why it ends up using data the way it does to make decisions. Several socially conscious computer and data scientists have recently started wrestling with the problem of machine bias. Some have come up with ways to add fairness requirements into machine-learning systems. Others have found ways to illuminate the sources of algorithms’ biased behavior. But the very nature of machine-learning algorithms as self-taught systems means there’s no easy fix to make them play fair.
Learning by example In most cases, machine learning is a game of algorithm see, algorithm do. The programmer assigns an algorithm a goal — say, predicting whether people will default on loans. But the machine gets no explicit instructions on how to achieve that goal. Instead, the programmer gives the algorithm a dataset to learn from, such as a cache of past loan applications labeled with whether the applicant defaulted.
The algorithm then tests various ways to combine loan application attributes to predict who will default. The program works through all of the applications in the dataset, fine-tuning its decision-making procedure along the way. Once fully trained, the algorithm should ideally be able to take any new loan application and accurately determine whether that person will default.
The trouble arises when training data are riddled with biases that an algorithm may incorporate into its decisions. For instance, if a human resources department’s hiring algorithm is trained on historical employment data from a time when men were favored over women, it may recommend hiring men more often than women. Or, if there were fewer female applicants in the past, then the algorithm has fewer examples of those applications to learn from, and it may not be as accurate at judging women’s applications. At first glance, the answer seems obvious: Remove any sensitive features, such as race or sex, from the training data. The problem is, there are many ostensibly nonsensitive aspects of a dataset that could play proxy for some sensitive feature. Zip code may be strongly related to race, college major to sex, health to socioeconomic status.
And it may be impossible to tell how different pieces of data — sensitive or otherwise — factor into an algorithm’s verdicts. Many machine-learning algorithms develop deliberative processes that involve so many thousands of complex steps that they’re impossible for people to review.
Creators of machine-learning systems “used to be able to look at the source code of our programs and understand how they work, but that era is long gone,” says Simon DeDeo, a cognitive scientist at Carnegie Mellon University in Pittsburgh. In many cases, neither an algorithm’s authors nor its users care how it works, as long as it works, he adds. “It’s like, ‘I don’t care how you made the food; it tastes good.’ ”
But in other cases, the inner workings of an algorithm could make the difference between someone getting parole, an executive position, a mortgage or even a scholarship. So computer and data scientists are coming up with creative ways to work around the black box status of machine-learning algorithms.
Setting algorithms straight Some researchers have suggested that training data could be edited before given to machine-learning programs so that the data are less likely to imbue algorithms with bias. In 2015, one group proposed testing data for potential bias by building a computer program that uses people’s nonsensitive features to predict their sensitive ones, like race or sex. If the program could do this with reasonable accuracy, the dataset’s sensitive and nonsensitive attributes were tightly connected, the researchers concluded. That tight connection was liable to train discriminatory machine-learning algorithms.
To fix bias-prone datasets, the scientists proposed altering the values of whatever nonsensitive elements their computer program had used to predict sensitive features. For instance, if their program had relied heavily on zip code to predict race, the researchers could assign fake values to more and more digits of people’s zip codes until they were no longer a useful predictor for race. The data could be used to train an algorithm clear of that bias — though there might be a tradeoff with accuracy.
On the flip side, other research groups have proposed de-biasing the outputs of already-trained machine-learning algorithms. In 2016 at the Conference on Neural Information Processing Systems in Barcelona, Hardt and colleagues recommended comparing a machine-learning algorithm’s past predictions with real-world outcomes to see if the algorithm was making mistakes equally for different demographics. This was meant to prevent situations like the one created by COMPAS, which made wrong predictions about black and white defendants at different rates. Among defendants who didn’t go on to commit more crimes, blacks were flagged by COMPAS as future criminals more often than whites. Among those who did break the law again, whites were more often mislabeled as low-risk for future criminal activity.
For a machine-learning algorithm that exhibits this kind of discrimination, Hardt’s team suggested switching some of the program’s past decisions until each demographic gets erroneous outputs at the same rate. Then, that amount of output muddling, a sort of correction, could be applied to future verdicts to ensure continued even-handedness. One limitation, Hardt points out, is that it may take a while to collect a sufficient stockpile of actual outcomes to compare with the algorithm’s predictions. A third camp of researchers has written fairness guidelines into the machine-learning algorithms themselves. The idea is that when people let an algorithm loose on a training dataset, they don’t just give the software the goal of making accurate decisions. The programmers also tell the algorithm that its outputs must meet some certain standard of fairness, so it should design its decision-making procedure accordingly.
In April, computer scientist Bilal Zafar of the Max Planck Institute for Software Systems in Kaiserslautern, Germany, and colleagues proposed that developers add instructions to machine-learning algorithms to ensure they dole out errors to different demographics at equal rates — the same type of requirement Hardt’s team set. This technique, presented in Perth, Australia, at the International World Wide Web Conference, requires that the training data have information about whether the examples in the dataset were actually good or bad decisions. For something like stop-and-frisk data, where it’s known whether a frisked person actually had a weapon, the approach works. Developers could add code to their program that tells it to account for past wrongful stops.
Zafar and colleagues tested their technique by designing a crime-predicting machine-learning algorithm with specific nondiscrimination instructions. The researchers trained their algorithm on a dataset containing criminal profiles and whether those people actually reoffended. By forcing their algorithm to be a more equal opportunity error-maker, the researchers were able to reduce the difference between how often blacks and whites who didn’t recommit were wrongly classified as being likely to do so: The fraction of people that COMPAS mislabeled as future criminals was about 45 percent for blacks and 23 percent for whites. In the researchers’ new algorithm, misclassification of blacks dropped to 26 percent and held at 23 percent for whites.
These are just a few recent additions to a small, but expanding, toolbox of techniques for forcing fairness on machine-learning systems. But how these algorithmic fix-its stack up against one another is an open question since many of them use different standards of fairness. Some require algorithms to give members of different populations certain results at about the same rate. Others tell an algorithm to accurately classify or misclassify different groups at the same rate. Still others work with definitions of individual fairness that require algorithms to treat people who are similar barring one sensitive feature similarly. To complicate matters, recent research has shown that, in some cases, meeting more than one fairness criterion at once can be impossible.
“We have to think about forms of unfairness that we may want to eliminate, rather than hoping for a system that is absolutely fair in every possible dimension,” says Anupam Datta, a computer scientist at Carnegie Mellon.
Still, those who don’t want to commit to one standard of fairness can perform de-biasing procedures after the fact to see whether outputs change, Hardt says, which could be a warning sign of algorithmic bias.
Show your work But even if someone discovered that an algorithm fell short of some fairness standard, that wouldn’t necessarily mean the program needed to be changed, Datta says. He imagines a scenario in which a credit-classifying algorithm might give favorable results to some races more than others. If the algorithm based its decisions on race or some race-related variable like zip code that shouldn’t affect credit scoring, that would be a problem. But what if the algorithm’s scores relied heavily on debt-to-income ratio, which may also be associated with race? “We may want to allow that,” Datta says, since debt-to-income ratio is a feature directly relevant to credit.
Of course, users can’t easily judge an algorithm’s fairness on these finer points when its reasoning is a total black box. So computer scientists have to find indirect ways to discern what machine-learning systems are up to.
One technique for interrogating algorithms, proposed by Datta and colleagues in 2016 in San Jose, Calif., at the IEEE Symposium on Security and Privacy, involves altering the inputs of an algorithm and observing how that affects the outputs. “Let’s say I’m interested in understanding the influence of my age on this decision, or my gender on this decision,” Datta says. “Then I might be interested in asking, ‘What if I had a clone that was identical to me, but the gender was flipped? Would the outcome be different or not?’ ” In this way, the researchers could determine how much individual features or groups of features affect an algorithm’s judgments. Users performing this kind of auditing could decide for themselves whether the algorithm’s use of data was cause for concern. Of course, if the code’s behavior is deemed unacceptable, there’s still the question of what to do about it. There’s no “So your algorithm is biased, now what?” instruction manual. The effort to curb machine bias is still in its nascent stages. “I’m not aware of any system either identifying or resolving discrimination that’s actively deployed in any application,” says Nathan Srebro, a computer scientist at the University of Chicago. “Right now, it’s mostly trying to figure things out.”
Computer scientist Suresh Venkatasubramanian agrees. “Every research area has to go through this exploration phase,” he says, “where we may have only very preliminary and half-baked answers, but the questions are interesting.”
Still, Venkatasubramanian, of the University of Utah in Salt Lake City, is optimistic about the future of this important corner of computer and data science. “For a couple of years now … the cadence of the debate has gone something like this: ‘Algorithms are awesome, we should use them everywhere. Oh no, algorithms are not awesome, here are their problems,’ ” he says. But now, at least, people have started proposing solutions, and weighing the various benefits and limitations of those ideas. So, he says, “we’re not freaking out as much.”
Here are the final images from Cassini’s last look around the Saturn system.
In its last hours before plunging into Saturn’s atmosphere, the Cassini spacecraft turned its cameras to the mission team’s favorite objects: the hydrocarbon-shrouded moon Titan, the geyser moon Enceladus and, of course, the majestic rings.
After sending these raw images back to Earth, Cassini reconfigured itself to stream data efficiently in near–real time. Image files are too big to send in this mode, so these are the last pictures Cassini will ever show us. But it will send back unprecedented information about Saturn’s atmosphere right up until the end.
The tiny moon Enceladus, which has a liquid sea below its icy surface and spews geysers of water into space, set behind Saturn as Cassini watched: Saturn looms large in this Sept. 14 raw image from the Cassini spacecraft: The hazy moon Titan is the largest in the Saturn system. Its gravity nudged Cassini onto its doomed orbit when the spacecraft flew by on September 11: